There are two main approaches to protein identification by the use of mass spectrometry:
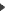 simple mass spectrometry (MS) ; simple mass spectrometry (MS) ;
tandem mass spectrometry (MS/MS).
Both approaches need the protein under study to be previously digested by a specific enzyme (usually trypsine).
MS analysis consists in weighing (by the use of "TOF" instruments - Time Of Flight measurements) each of the trypsin ions previously obtained. This results in a mass spectrum where every peaks represent an ion. Such a spectrum represents a fingerprint of the protein under study.
In MS/MS approach, each ion of the MS spetrum is fragmented and analysed in a second part of the mass spectrometer. This type of fragmentation (Y ions) occurs on specific positions of the trypsin fragment : the peptide bounds. As a result this method leads to the determination of the amino acid sequence of peptides.
 MS/MS analysis of the peptide fragment GLLGSLAGPK. MS/MS analysis of the peptide fragment GLLGSLAGPK.
Y ions allow the determination of the peptide sequence.
This example shows a partial MS/MS interpretation. |
All of the present activities of Helix and LCP/CEA in computational proteomics are based on this second approach: tandem mass spectrometry (MS/MS). |