La comparaison de séquences génomiques et protéiques est de loin la tâche informatique la plus fréquemment exécutée par les biologistes. Des algorithmes sont mis en uvre pour calculer les meilleurs alignements entre deux séquences.

Le principe de l'un de ces algorithmes, conçu par S. Needleman et C. Wunsch (J. Mol. Biol., 48, 443, 1970), est expliqué dans l'article « Séquences génétiques en face à face » du numéro 409 de juin 2007 de La Recherche (rubrique « Pas si simple - Wxyz », pages 82-83).
Son fonctionnement est détaillé ici à l'aide d'une applet Java.
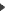 Sélectionnez tout d'abord la nature des séquences à aligner : génomiques ou protéiques. Sélectionnez tout d'abord la nature des séquences à aligner : génomiques ou protéiques.
Le coût d'une mutation peut être différent suivant la paire de caractères en jeu. Ces coûts sont donc fournis par une matrice 4 x 4 dans le cas des séquences génomiques (vous avez le choix entre la matrice unité et une matrice distinguant les coûts de transition et de réversion) ou par une matrice 20 x 20 dans le cas des séquences protéiques (seule la matrice PAM250 est disponible).
Enfin, vous devez assigner un coût à l'insertion d'un gap.
Vous pouvez alors lancer l'applet, qui, en 5 étapes listées dans le cadre « Etapes » en haut et à gauche de la fenêtre, vous aidera à comprendre pas à pas le principe de l'algorithme de Needleman et Wunsch.
À chaque étape, prenez connaissance des instructions et des messages qui sont affichés dans le cadre en bas à droite dans la fenêtre.
Attention : Le fonctionnement de ce programme requiert le plug'in Java2.
| Choix des séquences et de la matrice de substitution | | |
Ce programme a été développé par Régis Monte. |