Tandem mass spectrometry (MS/MS) presently represents the far most effective tool for high throughput proteomic analysis. MS/MS spectra provide informations that allow the interpretation of the sequence part of the proteins under study, leading to protein identifications.
In this context, Helix and the "Laboratoire Chimie des Protéines", with GENOME express for the PepMap module, designed PepLine: a software pipeline dedicated to high throughput proteomics. If most of the proteomic softwares achieve protein identification through requests on protein databases, PepLine offers moreover to localize genomic regions that putatively code for observed proteins that have not been discovered yet.
At the present time, PepLine is made up of two components: Taggor and PepMap.
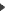 The Taggor component allows the partial sequence interpretation of the results produced by MS/MS experiments (MS/MS spectra peak lists) without any reconstruction phase against protein databases. Taggor output is a set of Peptide Sequence Tags. The Taggor component allows the partial sequence interpretation of the results produced by MS/MS experiments (MS/MS spectra peak lists) without any reconstruction phase against protein databases. Taggor output is a set of Peptide Sequence Tags.
A set of PSTs represents the input of the PepMap software. PepMap aims at identifying proteins through protein database screening or at localizing genes on a complete unannotated genome of the organism under study (FastA files).
These two software modules have been written in C language. Their validation on experimental data is in progress.
A first prototype of a graphic user interface dedicated to visualization and management of PepMap results has been implemented in Java.
 PepMap GUI PepMap GUIGene localization on the genome of Arabidopsis thaliana. The left panel of the PepMap GUI main window presents the PST best locations. The right panel displays the genomic location selected in the left panel (7 PSTs found). The optional window shows the nucleic sequence corresponding to the selected location (the nucleotides colored in red/green point out PST sequence/mass correspondances). | |